Prader-Willi Syndrome
Prader-Willi syndrome (PWS) is a complex disorder seen in association with mental retardation. Their IQ is usually between 60-70. PWS occurs between 10,000-1/15,000 live births. Descriptions of this disorder have been made for hundreds of years. Clinical features of PWS include hypotonia and failure to thrive in the newborn period (Figure 1). As the child grows obesity is present by age 6 unless actively avoided. Classical facial features include almond shaped eyes, long philtrum, cryptoorchism in males and small hands and feet. They have a unique behavioral phenotype. Behavioral features include skin picking, stubbornness, food hoarding and obsessive-compulsive behaviors.
PWS is caused by a deletion of the 15q11 region in 70% of patients (Figure 2). This deletion is of paternal origin. The critical gene for PWS is only active in the paternal chromosome 15. The maternal copy is methylated off. Methlyation studies looking at the 15q11 region is a molecular method useful in diagnosing PWS (figure 3). Patients with PWS have only a maternal pattern. This is a result of either deletion of the paternal copy of the chromosome 15 or both copies of the chromosome 15 being of maternal origin (uniparental disomy).
Management of Prader-willi includes treatment of their behavior and obesity. Growth hormone has been shown to improve obesity and increase height. When a patient has severe obesity they are at risk of sleep apnea. Growth hormone can be dangerous. Low fat diet and close monitoring of food is essential in management.
Angelman syndrome
This disorder occurs in 1/12,000 individuals. The children are often normal at birth. Developmental delay is often noted by 6-12 months. Ataxia and balance problems are often common. Children with Angelman syndrome have little or no language development. They often have outbursts of laughter, apparent happy demeanor, hand flapping and a short attention span. Clinical features include light pigment, broad jaw and tongue thrusting (figure 4).
Deletion of the 15q11 region is present in 70-75% of patients. In Angelman syndrome the deletion is of maternal origin. Mutations within the UBE3A gene are the cause in 20-25% of patients. Approximately 10% of patients with the clinical features of Angelman syndrome have no cytogenetic or molecular cause identified.
Management: Speech and language is significantly impaired in all patients. Nonverbal communication methods need to be emphasized. Patients with AS often place nonfood items in their mouth. The majority of patients with AS have seizures (90%). These can be difficult to control. Sleep disorders are also common. Children with AS appear to have a decreased need for sleep.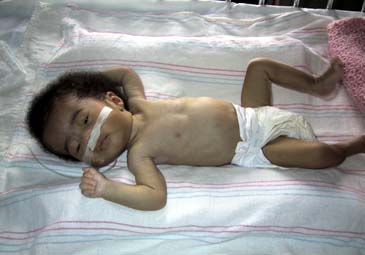
Figure 1.
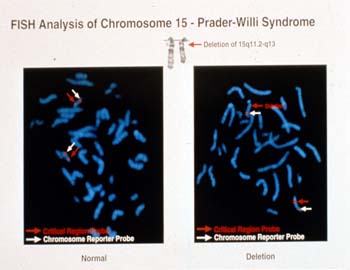
Figure 2.
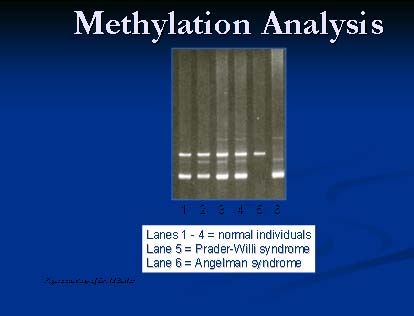
Figure 3.

Figure 4.
A 4 week old infant is noted to have hypotonia and failure to thrive. Chromosome analysis indicates a deletion of the 15q11 region.
- What is the diagnosis?
- What additional study is needed?
Methlyation studies indicate a maternal pattern.
- What medical complications is this child at risk for?
- If the methylation pattern indicated only paternal region present, would this change the diagnosis?